Several month ago, I was cruising on the packages folder of my Rstudio when I “accidentally” clicked on the “ape” package. At first, I saw nothing interesting….
Turns out, “ape” happened to be an acronym for Analysis of Phylogenetic and Evolution (how creative). Of course, that meant nothing to me, but I decided to look up what this was. Soon after, I ran into this.
{kind=link}

If you want to read up on more detail about this event, click here.
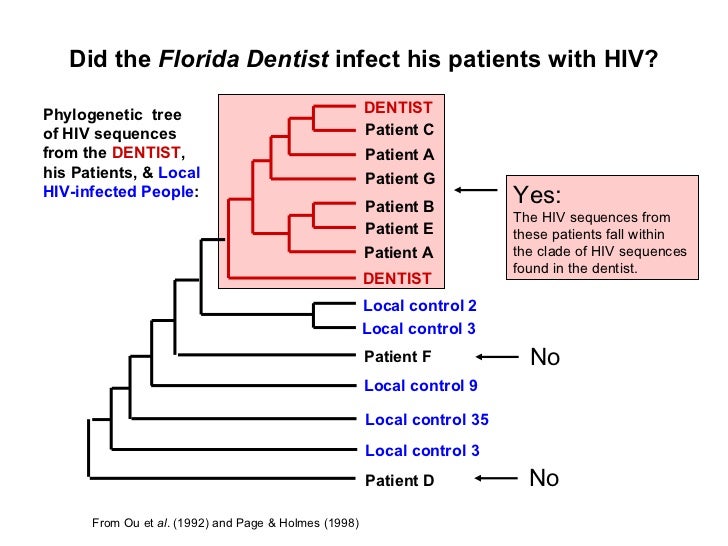
It turns out that this analysis was performed by laying out the viral traits of each patient and creating an analysis of correlation through a maximum likelihood sampling with replacement (bootstrap procedure). Although the technique depends on the assumption of independence of the original sample, the bootstrap can be used to estimate bias and variance for confidence intervals, and for hypothesis testing in many situations. After sampling, ordinal values are created based on the correlation of each attribute and distributed amongst the clades (branches) of the tree. That was a mouthful.
Simply said, this analysis takes like values and groups them together based on how similar they are. Essentially, this is a categorical version of a recurisvely partitioned tree model. As for the case of the dentist infecting HIV to the patient, samples of the blood were taken from 6 of the patients and compared with each other (along with other control groups) and found that the characteristics of the virus in the patients and the dentist correlated very closely in the same branch. This type of analysis may be useful in areas such as linguistics, evolutionary biology, and even detective work.
Can we say for certain that the doctor did malignantly infect his patients? No. Yet, there is potential evidence!
After reading all this, I decided that I wanted to make my own tree diagram even though I knew nothing about how to go about it. After realizing how difficult this task was going to be, I resorted to create just one tree that simply showed how closely linked a group of interest was. I am sure that I can explore other packages. Someone is bound to have already created something similar.
Project
So… Let’s create our own phylogenetic trees.
Just for fun, we have a csv of a supposed “top ten” coding languages. 14 of the points were collected randomly on the internet with the 10th language included as R. The .csv is at the bottom for reference. We first start out by installing the “ape” package and creating a function to read the data.
library("ape")
# Set up original functions
readData <- function(path) {
table <- read.csv(path, stringsAsFactors=FALSE)
return(table)
} #1st
Next, is a function that takes the table and creates a matrix that returns the correlating values of each row. Probably could have done it simpler, but it was just hard-coded.
Relationship <- function(table) {
rows <- nrow(table)
cols <- ncol(table)
rMatrix <- matrix(0,nrow = rows, ncol = rows)
for (i in 1:(rows - 1))
for (j in (i+1):rows)
for (k in 2:cols)
if (table[i,k] == table[j,k]) {
rMatrix[i,j] <- rMatrix[i,j] + 1
# May adjust to add weighted value
return(rMatrix)}
}
Taking inputs of our csv and the relationship matrix, we will create labels and match each part of the tree. The correlation matrix is transformed and attached to connect ordinal values. Now it will match the “Newick” or “New Hampshire” format.
makeTreeText <- function(matrix, table) { # Longer function
# offset function used to take two values and take the
# first value - 1 unless the second is bigger,
# then return second value.
offset <- function(val, vec) {
offset <- 0
for(item in vec) {
if (val > item)
offset <- offset + 1
}
return(val - offset)
}
# function creates a new matrix
averageMatrix <- function(matrix, vec) {
newMatrix <- matrix(0, nrow=nrow(matrix) - (length(vec) - 1),
ncol=ncol(matrix) - (length(vec) - 1))
combine <- vec[1]
remove <- vec[2:length(vec)]
for (col in 2:ncol(matrix)) {
for (row in 1:(col-1)) {
if ((row %in% remove) || (col %in% remove)){
next
}
else if (row == combine) {
newCol <- offset(col, remove)
sum <- matrix[row,col]
for(r in remove) {
sum <- sum + matrix[min(r,col), max(r,col)]
}
newMatrix[row,newCol] <- sum/length(vec)
}
else if (col == combine){
newRow <- offset(row, remove)
sum <- matrix[row,col]
for(c in remove) {
sum <- sum + matrix[min(c,row), max(c,row)]
}
newMatrix[newRow,col] <- sum/length(vec)
}
else {
newRow <- offset(row, remove)
newCol <- offset(col, remove)
newMatrix[newRow, newCol] <- matrix[row,col]
}
}
}
return(newMatrix)
}
# These titles are merged to pass through the plot Treefunction!
mTitles <- function(titles, indices) {
merged <- paste(titles[indices], collapse=",")
merged <- paste("(",merged,")", sep=" ")
remove <- titles[indices[2:length(indices)]]
dTitles <- titles[!titles %in% remove]
dTitles[indices[1]] <- merged
return(dTitles)
}
# Just a find and choose function
uniqueAppend <- function(vec, val) {
if(val %in% vec)
return(vec)
return(append(vec, val))
}
# Finally combining the titles with the new average matrix
text <- ""
titles <- table[,1]
while(length(titles) > 1) {
highVal <- 0
highVec <- c()
for (col in 2:ncol(matrix)) {
for (row in 1:(col-1)) {
if(matrix[row, col] > highVal) {
highVal <- matrix[row, col]
highVec <- c(row, col)
}
else if(matrix[row,col] == highVal &&
(row %in% highVec || col %in% highVec)) {
highVec <- uniqueAppend(highVec, row)
highVec <- uniqueAppend(highVec, col)
}
}
}
titles <- mTitles(titles, highVec)
matrix <- averageMatrix(matrix, highVec)
}
text <- titles[1]
text <- paste(text,";", sep=" ")
return(text)
Finally, we are combining the functions and plotting the tree. Using the read.tree() function.
# PLOT TREE FUNCTION BELOW
plotTree <- function(path) {
table <- readData(path)
matrix <- buildRelationshipMatrix(table)
phylogenyText <- makeTreeText(matrix,table)
tree <- read.tree(text=phylogenyText)
plot(tree)
}
plotTree("prog.csv")

All that for a simple tree.
This is a brute-force recursive partitioned tree. Much like what biologists use today, they figure what variables carry the most weight and separate the values according to the various explanatory factors.
This tree will work with as many categorical values. If you would like to give this example csv a try, give it a go!
Conclusion
This is less of an analysis post but more of a coding project that shows the power R has to be just as object-oriented as any other language. Of course, R is still nothing in comparison to the speed of languages such as C++ and python. Soon, it will catch up in other ways.
download the original matrix
download an example matrix